 |
Nanoprobes, Incorporated
95 Horseblock Road, Unit 1, Yaphank, NY 11980-9710
Tel: (877) 447-6266 (Toll-Free in US) or (631) 205-9490 | Fax: (631) 205-9493
tech@nanoprobes.com | www.nanoprobes.com |
Updated: March 26, 2000
NANOGOLD® PRODUCT INFORMATION
Monoamino Nanogold® Labeling Reagent
[Monoamino Nanogold® Labeling Reagent product page]

| Product Name: |
MONOAMINO NANOGOLD IN 5 ALIQUOTS |
| Catalog Number: |
2021A |
| Appearance: |
Brown solid |
| Revision: |
1.4 (March 2000) |
Technical Assistance Online
![[2021A-PDF]](../Images/pdf.gif) Instructions (PDF) Instructions (PDF)
Material Safety Data Sheet (PDF)
Congratulations on your acquisition of a revolutionary new gold immunoreagent: the MONOAMINO NANOGOLD® labeling kit. With this reagent you can label your own glycoproteins, other biomolecules and probes containing aldehydes or other reactive functional groups, with NANOGOLD®. Because NANOGOLD® is a discrete molecular compound and not a colloidal gold preparation, conjugates prepared with this reagent have several advantages over colloidal gold conjugates (see below).
Guide to Gold Cluster Labeling: Our detailed description of gold cluster labeling, how to isolate conjugates and calculate the extent of labeling, is now available, including spectra, micrographs, and tips and suggestions for better labeling (opens in a new window).
Warning: For research use only. Not recommended or intended for diagnosis of disease in humans or animals. Do not use internally or externally in humans or animals. Non radioactive and non carcinogenic.
NANOGOLD® is a newly developed gold label, prepared using a discrete gold compound rather than a colloid.1 This kit contains the NANOGOLD® particle with a single primary amine functionality incorporated into a ligand on the surface of the gold particle; this has a specific reactivity towards aldehyde groups, with which it forms Schiff bases. These may then be reduced to amines. This process may be used to label carbohydrate moities in glycoproteins,2,3 as shown below (figure 1). It may also be used with cross-linking reagents to label other groups, such as primary amines.4 The reagent as supplied has been lyophilized from methanol; dissolution in the reaction solvent or buffer will produce a solution of activated NANOGOLD®. NANOGOLD® conjugates can be used in immunoblotting, light microscopy, and electron microscopy to provide clear visibility. They are stable to wide ranges of pH and ionic strength, and are not radioactive or carcinogenic. The labeling reagent should be stored at -20°C.
Contents
MONOAMINO NANOGOLD® is supplied lyophilized from methanol solution. The dark brown solid may be dissolved in aqueous buffer systems such as phosphate buffered saline or ammonium acetate, and is also soluble in alcohols, acetone, DMSO, dichloromethane and other organic solvents. Extinction coefficients at specific wavelengths are given below for methanol solution:
| WAVELENGTH (nm) |
EXTINCTION COEFFICIENT* |
| 280 |
2.25 X 105 |
| 420 |
1.12 X 105 |
| |
*Measured for 5 X 10-6 M solution in methanol. |
5 X 6 nmol of the product is supplied. The amounts required will vary according to the system under study, but in general a 6-fold to 10-fold excess of NANOGOLD® is recommended; the reagent supplied will therefore label up to 0.1 mg of a molecule with one labeling site and a molecular weight of 100,000.
Contents
NANOGOLD® particles are not stable above 50°C. Best results are obtained at room temperature or 4°C. Avoid 37°C incubations. Use low temperature embedding media (e.g., Lowicryl) if labeling before embedding; do not bake tissue blocks with NANOGOLD®.
NANOGOLD® particles degrade upon exposure to thiols such as beta-mercaptoethanol or dithiothreitol. Thiol compounds used in the reduction of protein molecules (or other biomolecules) should be separated from the protein by gel filtration chromatography before NANOGOLD® conjugation. Use a gel, such as Amicon GH-25, which has an exclusion cut-off at molecular weight 3,000. Dialysis does NOT provide acceptable purification in this application. A small amount of residual thiol reagent can severely limit the performance of NANOGOLD®.
Contents
MONOAMINE NANOGOLD® is reactive towards aldehydes. It may be used to label carbohydrate moieties of glycoproteins2 as shown in figure 1.
Sufficient NANOGOLD® reagent is supplied to label up to 1 nmol of aldehyde groups (for example, 0.1 mg of a compound with one aldehyde and a molecular weight of 100,000). Once the NANOGOLD® is dissolved in aqueous solution it is stable for a few days to several weeks, with higher ionic strengths favoring longer stability. For purification and isolation steps alternative buffers may be substituted for those given; however, the labeling reaction itself should be performed using the buffers and conditions specified.
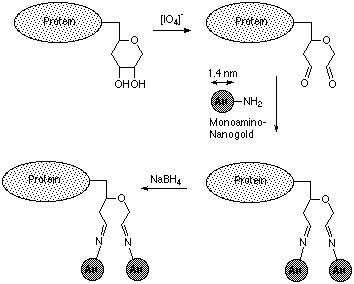
Figure 1: Labeling of a carbohydrate moiety with MONOAMINO NANOGOLD® (Ref. 2)
To assist structural preservation, it is usually helpful to fix proteins before labeling. If glutaraldehyde or other aldehyde-containing fixing reagents are used, these should be quenched before labeling. This may be achieved by reacting with sodium borohydride (15-20 mole equivalents per glutaraldehyde) at 4°C for 1.5 h (Ref. 2) Alternatively, incubate the specimens for 5 minutes in 50 mM glycine solution in PBS (pH 7.4), or treat with ammonium chloride (50 mM).
The procedure below is a recent modification of our original procedure which has been found to give more selective labeling.3 Sugars must be oxidized to aldehydes for reaction with MONOAMINO NANOGOLD® to take place. Sodium periodate at 1 mM, 0°C slectively cleaves only sialic acid. At 10 mM or higher concentrations, other sugars are cleaved. The concentration of the periodate used should be adjusted accordingly.
Oxidation:
- Dissolve the glycoprotein in 0.01 - 0.1 M phosphate buffer, pH 7.0. Other buffers and pH values are acceptable, except that amine-containing bufers such as tris or glycine must be avoided.
- Make a 10 mg/mL solution of sodium periodate in water, protect from light. Add to the protein to make a final concentration of 10 mM periodate (1 mM if labeling at sialic acid); protect from light.
- React at room temperature for 15 - 30 minutes.
- Sepatate the oxidized glycoprotein from excess periodate by gel filtration; optionally, the unreacted periodate may be quenched beforehand with 0.1 mL glycerol/mL of reaction mixture.
Alternatively, enzymes may be used to oxidize the diol groups (e.g. glucose or galactose oxidases). An example is given below - oxidase reaction for cell labeling of galactose.
Enzymatic oxidation:
- Prepare 5 % cell suspension in the appropriate buffer. Avoid amine-containing buffers such as tris or glycine.
- Add 0.05 units of vibrio cholerae neuraminidase (to remove sialic acid sugars to expose galactose as the terminal residue) and 5 units of galactose oxidase per mL of cell suspension.
- Incubate for 60 minutes at 37°C.
Gold labeling:
- Dissolve oxidized protein in 0.2 M sodium carbonate buffer, pH 9.6, at a concentration of 10 mg/mL.
- Dissolve MONOAMINO NANOGOLD® in the same buffer. Use a 5 to 15-fold stoichiometric excess over the groups to be labeled.
- Mix and allow to react at room temperature for 2 hours.
- Add 10 microliters of 5 M sodium cyanoborohydride per mL of reaction (CAUTION: Toxic - do this in a fume hood). The 5 M solution is prepared in 1 N sodium hydroxide.
- React for 30 minutes at room temperature.
- Block unreacted aldehyde with 50 microliters of 1 M ethanolamine, pH 9.6 per mL of reaction mixture. A 1 M ethanolamine solution is prepared by the addition of 300 microliters of ethanolamine to 5 mL of water and adjusting the pH with concentrated hydrochlorid acid while on ice.
- React for 30 minutes at room temperature.
- Separate the NANOGOLD®-labeled conjugate by gel filtration, eluting with 10 mM sodium phosphate buffer with 0.15 M sodium chloride, pH 7.0. Other buffers and pH values are acceptable.
The conventional procedure is given below:
- If the labeling site is in the form of a carbohydrate group, it must be oxidized to produce aldehydes. Incubate specimen for 1.5 h at 4°C with 1000-2000 mole equivalents of sodium periodate per carbohydrate in 20 mM PIPES.Na.
- Isolate oxidized glycoprotein by by dialysis or gel filtration chromatography to remove sodium periodate. Use a gel such as Amicon GH-25, which has an exclusion cut-off at molecular weight 3,000. As the eluent, use 20mM PIPES.Na, with 150 mM sodium chloride. The oxidizd glycoprotein will be eluted in the void volume as the first sharp peak in the trace. Combine the fractions containing protein; the total amount may be calculated from the optical density.
- Dissolve the NANOGOLD® reagent in 1 ml buffer. Solution is facilitated by first dissolving the reagent in 0.02 ml isopropanol, then diluting to 0.2 ml with the buffer used for the reaction. Sufficient reagent is supplied to label up to 1 nmol of aldehydes; if you are using a smaller amount, use a proportionately smaller amount of the NANOGOLD® reagent. Once activated NANOGOLD® is reconstituted with water it should be used within one week.
- Add the MONOAMINO NANOGOLD® solution to the oxidized glycoprotein (6 to 10 mole equivalents percarbohydrate chain). Incubate for 1 hour at 4°C.
- Reduce the Schiff base linkages with excess NaBH4; use 500-1000 mole equivalents per carbohydrate chain, and allow to react for 30 minutes at 4°C; then quench with excess acetone.
- Separate the unbound gold particles from the labeled glycoprotein using gel exclusion chromatography. The NANOGOLD® conjugate may be effectively isolated using a medium such as Pharmacia Superose 6 or 12 (which fractionate a wide range of molecular weights) or Amicon GCL-90 (which excludes molecules of mass 30,000 or greater). Concentrate the reaction mixture to a suitably small volume using membrane centrifugation (e.g. Amicon Centricon-30 system). Elute with 0.25 M ammonium acetate (nominal pH 7.7); monitor at 280 nm. The first, faintly colored peak is the conjugate, while the second, darker band is unbound NANOGOLD® particles. For even higher purity, repeat this process one time.
NANOGOLD® conjugates should be stored in 0.02 M phosphate buffer buffer with 150 mM sodium chloride, or other buffer solutions usually used with the protein under study. If they are to be stored longer than three days, add 0,1 % bovine serum albumin and 0.05 % sodium azide to prevent bacterial contamination and to prevent the protein from adhering to the surfaces of the storage vessel.
If the molecule to be labeled already contains aldehydes, the procedure above should be followed without the periodate oxidation and removal.
Contents
MONOAMINO NANOGOLD® may be used to label other groups if it is used with a suitable cross-linker. As an example, the use of bis (sulfosuccinimidyl) suberate (BS3)as a cross linker to label a protein with a primary amine is described below.4 The reaction works best in concentrated protein solutions (i.e. smaller reaction volumes).
- Dissolve the protein in 0.02 M sodium phosphate with 150 mM sodium chloride at pH 7.4 (0.5 ml).
- Dissolve the NANOGOLD® reagent in 0.2 ml buffer. Solution is facilitated by first dissolving the reagent in 0.02 ml isopropanol, then diluting to 0.2 ml with 0.02 M phosphate buffer with 150 mM NaCl, pH 7.4. Sufficient reagent is supplied to label up to 1 nmol of sites; if you are using a smaller amount, use a proportionately smaller amount of NANOGOLD® reagent. Once activated NANOGOLD® is reconstituted with water it should be used within one week.
- Add the MONOAMINO NANOGOLD® solution to the protein (6 to 10 mole equivalents per mole equivalent amine labeling site). Mix thoroughly.
- Dissolve BS3 in DMSO (0.1 ml) and dilute to 2 ml with 0.02 M phosphate buffer with 150 mM NACl, pH 7.4. Use sufficient BS3 to give a concentration of 1-2 mM in the reaction mixture (0.5 mg). Once dissolved, the cross-linking reagent should be used immediately. Hydrolysis will occur within a few hours.
- Add the BS3 solution to the protein/NANOGOLD® solution, and incubate for 10 mins at room temperature.
- Quench with excess 10 mM tris buffer at pH 7.0.
- Separate the unbound gold particles from the labeled glycoprotein using gel exclusion chromatography. The NANOGOLD® conjugate may be effectively isolated using a medium such as Pharmacia Superose 6 or 12 (which fractionate a wide range of molecular weights) or Amicon GCL-90 (which excludes molecules of mass 30,000 or greater). Concentrate the reaction mixture to a suitably small volume using membrane centrifugation (e.g. Amicon Centricon-30 system). Elute with 0.25 M ammonium acetate (nominal pH 7.7); monitor at 280 nm. The first, faintly colored peak is the conjugate, while the second, darker band is unbound NANOGOLD® particles. For even higher purity, repeat this process one time.
The extent of labeling may be determined from the UV/visible spectrum of the conjugate. NANOGOLD® has an extinction coefficient at 280 nm of 2.25 X 105, and at 420 nm of 1.12 X 105.
Other cross-linking reagents may be used to link the MONOAMINO NANOGOLD® to other functionalities. The buffer and reagent concentrations used for this reaction, and the incubation time, may be varied according to the nature of the molecule under study.
Contents
Basically, normal methodologies may be used successfully with NANOGOLD® labeled proteins. If labeling is successful, then the nature of the protein should be little affected.
The major difference will be in the results:
- NANOGOLD® is an extremely uniform 1.4 nm diameter gold particle (ca.10%).
- NANOGOLD® conjugates contain absolutely no aggregates. This is in sharp contrast to other colloidal gold conjugates that usually are prepared by centrifugation to remove the largest aggregates and frequently contain smaller aggregates.
- Close to 1 NANOGOLD® particle to 1 labeling site make this product distinct from the 0.2 - 10 variable stoichiometry of other colloidal gold antibody preparations.
- NANOGOLD® particles do not have affinity to proteins as do other other colloidal golds. This reduces background and false labeling.
- NANOGOLD® develops better with silver than do most other colloidal golds giving it higher sensitivity. Silver enhancement can be used to make the immunolabeling useful for electron microscopy, light microscopy, and immunoblotting with improved results.
Contents
Because the 1.4 nm NANOGOLD® particles are so small, over staining with OsO4, uranyl acetate or lead citrate may tend to obscure direct visualization of individual NANOGOLD® particles. Three recommendations for improved visibility of NANOGOLD® are:
- Use of reduced amounts or concentrations of usual stains.
- Use of lower atomic number stains such as NANOVAN, a Vanadium based stain.7
- Enhancement of NANOGOLD® with silver developers, such as LI SILVER or HQ SILVER.
Contents
Procedures are suggested below for the use of NANOGOLD® labeled proteins or other molecules as labeling reagents. NANOGOLD® labeled reagents are compatible with all commonly used aqueous buffer systems and pH values. Procedures, conditions and buffer systems may be changed or unnecessary steps dropped according to the properties of the system to be studied.
Cells in Suspension
- Optional fixing of cells: e.g., with glutaraldehyde (0.05 - 1% for 15 minutes) in PBS. Do not use Tris buffer since this contains an amine. After fixation, centrifuge cells (e.g. 1 ml at 10,000,000 cells/ml) at 300 X g, 5 minutes; discard supernatant; resuspend in 1 ml buffer. Repeat this washing (centrifugation and resuspension) 2 times.
- Incubate cells with 0.02 M glycine in PBS (5 mins). Centrifuge, then resuspend cells in PBS-BSA buffer (specified below) for 5 minutes.
- Wash cells using PBS-BSA as described in step 1 (2 X 5 mins). Resuspend in 1 ml Buffer 1.
- Place 50 - 200 ml of cells into Eppendorf tube. Dilute NANOGOLD® conjugate ~ 50 times in PBS-BSA buffer and add 30 ml to cells; incubate for 30 minutes with occasional shaking (do not create bubbles which will denature proteins).
- Wash cells in PBS-BSA as described in step 1 (2 X 5 mins).
- Fix cells and reagents using a final concentration of 1% glutaraldehyde in PBS for 15 minutes. Then remove fixative by washing with buffer 1 (3 X 5 mins).
| PBS-BSA Buffer:
20 mM phosphate
150 mM NaCl
pH 7.40
0.5% BSA
0.1% gelatin (high purity)
Optional, may reduce background:
0.5 M NaCl
0.05% Tween 20 |
PBS Buffer: 20 mM phosphate
150 mM NaCl
pH 7.40 |
Negative staining may be used for electron microscopy of small structures or single molecules which are not embedded. Negative stain must be applied after the silver enhancement. NANOVAN negative stain is specially formulated for use with NANOGOLD® reagents; it is based on vanadium, which gives a lighter stain than uranium, lead or tungsten-based negative stains and allows easier visualization of NANOGOLD® particles with little or no silver enhancement.
Thin Sections
Labeling with NANOGOLD® may be performed before or after embedding.5 Labeling before embedding and sectioning (the pre-embedding method) is used for the study of surface antigens, particularly small organisms such as viruses budding from host cells. It gives good preservation of cellular structure, and subsequent staining usually produces high contrast for study of the cellular details. Labeling after embedding and sectioning (the post-embedding method) allows the labeling protein access to the interior of the cells, and is used to label both exterior and interior features. The procedures for both methods are described below.
Thin sections mounted on grids are floated on drops of solutions on parafilm or in well plates. Hydrophobic resins usually require pre-etching.
CAUTION: NANOGOLD® particles are not stable above 37°C. If labeling is performed before embedding, an embedding medium should be used that hardens at low temperature (<30°C, e.g. Lowicryl). See temperature caution above.
PROCEDURE FOR PRE-EMBEDDING METHOD:5
- Float on a drop of water for 5 - 10 minutes.
- Incubate cells with 1 % bovine serum albumin in PBS buffer at pH 7.4 for 5 minutes; this blocks any non-specific protein binding sites and minimizes non-specific antibody binding.
- Rinse with PBS-BSA (1 min).
- Incubate with NANOGOLD® conjugate diluted 1/40 - 1/200 in PBS-BSA for 10 minutes to 1 hour at room temperature.
- Rinse with PBS-BSA (3 X 1 min), then PBS (3 X 1 min).
- Postfix with 1 % glutaraldehyde in PBS (10 mins).
- Rinse in deionized water (2 X 5 min).
- Dehydrate and embed according to usual procedure. Use of a low-temperature resin (eg. Lowicryl) is recommended.
- Stain (uranyl acetate, lead citrate or other positive staining reagent) as usual before examination.
Silver enhancement may be performed before or after embedding (see below); it should be completed before postfixing or staining with osmium tetroxide, uranyl acetate or similar reagents is carried out.
PROCEDURE FOR POST-EMBEDDING METHOD:5
- Prepare sections on plastic or carbon-coated nickel grid. Float on a drop of water for 5 - 10 minutes.
- Incubate with 1 % solution of bovine serum albumin in PBS buffer at pH 7.4 for 5 minutes to block non-specific protein binding sites.
- Rinse with PBS-BSA (1 min).
- Incubate with NANOGOLD® conjugate diluted 1/40 - 1/200 in PBS-BSA for 10 minutes to 1 hour at room temperature.
- Rinse with PBS (3 X 1 min).
- Postfix with 1 % glutaraldehyde in PBS at room temperature (3 mins).
- Rinse in deionized water for (2 X 5 min).
- If desired, contrast sections with uranyl acetate and/or lead citrate before examination.
Silver enhancement may also be used to render the NANOGOLD® particles more easily visible (see below), especially if stains such as uranyl acetate or lead citrate are applied. If used, it should be completed before these stains are applied.
| PBS-BSA Buffer:
20 mM phosphate
150 mM NaCl
pH 7.40
0.5% BSA
0.1% gelatin (high purity)
Optional, may reduce background:
0.5 M NaCl
0.05% Tween 20 |
PBS Buffer: 20 mM phosphate
150 mM NaCl
pH 7.40 |
Contents
For most work, silver enhancement is recommended to give a good signal in the electron microscope (see below). For particular applications, visualization of the NANOGOLD® directly may be desirable. Generally this requires very thin samples and precludes the use of other stains.
NANOGOLD® provides a much improved resolution and smaller probe size over colloidal gold conjugates. However, because NANOGOLD® is only 1.4 nm in diameter, it will not only be smaller, but will appear less intense than, for example, a 5 nm gold particle. With careful work, however, NANOGOLD® may be seen directly through the binoculars of a standard EM even in 80 nm thin sections. However, achieving the high resolution necessary for this work may require new demands on your equipment and technique. Several suggestions follow:
- Before you start a project with NANOGOLD® it is helpful to see it so you know what to look for. Dilute the NANOGOLD® stock 1:5 and apply 4 ml to a grid for 1 minute. Wick the drop and wash with deionized water 4 times.
- View NANOGOLD® at 100,000 X magnification with 10 X binoculars for a final magnification of 1,000,000 X. Turn the emission up full and adjust the condenser for maximum illumination.
- The alignment of the microscope should be in order to give 0.3 nm resolution. Although the scope should be well aligned, you may be able to skip this step if you do step 4
- Objective stigmators must be optimally set at 100,000 X. Even if the rest of the microscope optics are not perfectly aligned, adjustment of the objective stigmators may compensate and give the required resolution. You may want to follow your local protocol for this alignment but since it is important, a brief protocol is given here:
- At 100,000 X (1 X 106 with binoculars), over focus, under focus, then set the objective lens to in focus. This is where there is the least amount of detail seen.
- Adjust each objective stigmator to give the least amount of detail in the image.
- Repeat steps a and b until the in focus image contains virtually no contrast, no wormy details, and gives a flat featureless image.
- Now underfocus slightly, move to a fresh area, and you should see small black dots of 1.4 nm size. This is the NANOGOLD®. For the 1:5 dilution suggested, there should be about 5 to 10 gold spots on the small viewing screen used with the binoculars. Contrast and visibility of the gold clusters is best at 0.2 - 0.5 m defocus, and is much worse at typical defocus values of 1.5 - 2.0 m commonly used for protein molecular imaging.
- In order to operate at high magnification with high beam current, thin carbon film over fenestrated holey film is recommended. Alternatively, thin carbon or 0.2% Formvar over a 1000 mesh grid is acceptable. Many plastic supports are unstable under these conditions of high magnification/high beam current and carbon is therefore preferred. Contrast is best using thinner films and thinner sections.
- Once you have seen NANOGOLD® you may now be able to reduce the beam current and obtain better images on film. For direct viewing with the binoculars reduction in magnification from 1,000,000 X to 50,000 X makes the NANOGOLD® much more difficult to observe and not all of the golds are discernable. At 30,000 X (300,000 X with 10 X binoculars) NANOGOLD® particles are not visible. It is recommended to view at 1,000,000 X, with maximum beam current, align the objective stigmators, and then move to a fresh area, reduce the beam, and record on film.
- If the demands of high resolution are too taxing or your sample has an interfering stain, a very good result may be obtained using silver enhancement to give particles easily seen at lower magnification.
Contents
NANOGOLD® will nucleate silver deposition resulting in a dense particle 2-80 nm in size or larger depending on development time. If specimens are to be embedded, silver enhancement is usually performed after embedding, although it may be done first. It must be completed before any staining reagents such as osmium tetroxide, lead citrate or uranyl acetate are applied, since these will nucleate silver deposition in the same manner as gold and produce non-specific staining. With NANOGOLD® reagents, low-temperature resins (eg Lowicryl) should be used and the specimens kept at or below room temperature until after silver development has been completed. Silver development is recommended for applications of NANOGOLD® in which these stains are to be used, otherwise the NANOGOLD® particles may be difficult to visualize against the stain.
Our LI SILVER silver enhancement system is convenient and not light sensitive, and suitable for all applications. Improved results in the EM may be obtained using HQ SILVER, which is formulated to give slower, more controllable particle growth and uniform particle size distribution.
Specimens must be thoroughly rinsed with deionized water before silver enhancement reagents are applied. This is because the buffers used for incubations and washes contain chloride ions and other anions which form insoluble precipitates with silver. These are often light-sensitive and will give non-specific staining. To prepare the developer, mix equal amounts of the enhancer and initiator immediately before use. NANOGOLD® will nucleate silver deposition resulting in a dense particle 2-20 nm in size or larger depending on development time. Use nickel grids (not copper).
The procedure for immunolabeling should be followed up to step 6 as described above. Silver enhancement is then performed as follows:
- Rinse with deionized water (2 X 5 mins).
- OPTIONAL (may reduce background): Rinse with 0.02 M sodium citrate buffer, pH 7.0 (3 X 5 mins).
- Float grid with specimen on freshly mixed developer for 1-8 minutes, or as directed in the instructions for the silver reagent. More or less time can be used to control particle size. A series of different development times should be tried, to find the optimum time for your experiment. With HQ Silver, a development time of 6 min gives 15-40 nm round particles.
- Rinse with deionized water (3 X 1 min).
- Mount and stain as usual.
NOTE: Treatment with osmium tetroxide followed by uranyl acetate staining can lead to much more drastic loss of the silver enhanced NANOGOLD® particles. This may be prevented by gold toning:6
- After silver enhancement, wash thoroughly with dionized water.
- 0.05 % gold chloride: 10 minutes at 4°C.
- Wash with deionized water.
- 0.5 % oxalic acid: 2 mins at room temperature.
- 1 % sodium thiosulfate (freshly made) for 1 hour.
- Wash thoroughly with deionized water and embed according to usual procedure.
Contents
Features labeled with NANOGOLD® will be stained black in the light microscope upon silver enhancement. Different development times should be tried to determine which is best for your experiment. The procedure for immunolabeling is similar to that for EM; a suitable procedure is given below.
Samples must be rinsed with deionized water before silver enhancement. This is because the reagent contains silver ions in solution, which react to form a precipitate with chloride, phosphate and other anions which are components of buffer solutions. The procedure for immunolabeling with NANOGOLD® and silver enhancement is given below.
- Spin cells onto slides using Cytospin, or use parafin section.
- Incubate with 1 % solution of bovine serum albumin in PBS (PBS-BSA) for 10 minutes to block non-specific protein binding sites.
- Rinse with PBS-BSA (3 X 2 min).
- Incubate with NANOGOLD® conjugate diluted 1/40 - 1/200 in PBS-BSA for 1 hour at room temperature.
- Rinse with PBS (3 X 5 min).
- Postfix with 1 % glutaraldehyde in PBS at room temperature (3 mins).
- Rinse with deionized water (3 X 1 min).
- OPTIONAL (may reduce background): Rinse with 0.02 M sodium citrate buffer, pH 7.0 (3 X 5 mins).
- Develop specimen with freshly mixed developer for 5-20 minutes, or as directed in the instructions for the silver reagent. More or less time can be used to control intensity of signal. A series of different development times may be used, to find the optimum enhancement for your experiment; generally a shorter antibody incubation time will require a longer silver development time.
- Rinse with deionized water (2 X 5 mins).
- The specimen may now be stained if desired before examination, with usual reagents.
| PBS-BSA Buffer:
20 mM phosphate
150 mM NaCl
pH 7.40
0.5% BSA
0.1% gelatin (high purity)
Optional, may reduce background:
0.5 M NaCl
0.05% Tween 20 |
PBS Buffer: 20 mM phosphate
150 mM NaCl
pH 7.40 |
To obtain an especially dark silver signal, the silver enhancement may be repeated with a freshly mixed portion of developer.
Contents
The basic procedure for gold immunoblotting has been described by Moeremans et al,7 which may be followed. For best results, the membrane should be hydrated before use by simmering in gently boiling water for 15 minutes. Best results are obtained when the antigen is applied using a 1 microliter capillary tube. The procedure for blots is as follows, if the NANOGOLD® conjugate is the primary labeling reagent:
- Spot 1 microliter dilutions of the antigen in buffer 4 onto hydrated nitrocellulose membrane. Use an antigen concentration range from 100 ng to 0.01 pg / microliter.
- Block with buffer 1 for 30 minutes at 37°C.
- Rinse with buffer 1 (3 X 10 mins).
- Incubate with a 1/100 to 1/200 dilution of the NANOGOLD® reagent in buffer 2 for 2 hours at room temperature.
- Rinse with buffer 3 (3 X 5 mins), then buffer 4 (2 X 5 mins).
- OPTIONAL (may improve sensitivity): Postfix with glutaraldehyde, 1 % in buffer 4 (10 mins).
- Rinse with deionized water (2 X 5 mins).
- OPTIONAL (may reduce background): Rinse with 0.05 M EDTA at pH 4.5 (5 mins).
- Develop with freshly mixed silver developer for 20-25 minutes or as directed in the instructions for the silver reagent, twice. Rinse thoroughly with deionized water between developments to remove all the reagent.
- Rinse several times with deionized water.
| Buffer 1: 20 mM phosphate
150 mM NaCl
pH 7.40
4% BSA (bovine serum albumin)
2 mM sodium azide (NaN3) |
Buffer 2:
20 mM phosphate
150 mM NaCl
pH 7.4
0.8% BSA
1% normal serum; use serum of the host animal for the NANOGOLD® antibody
0.1% gelatin (Type B, approx. 60 bloom)
Optional, may reduce background:
0.5 M NaCl
0.05% Tween 20
|
| Buffer 3: 20 mM phosphate
150 mM NaCl
pH 7.40
0.8% BSA (bovine serum albumin)
2 mM sodium azide |
Buffer 4 (PBS): 20 mM phosphate
150 mM NaCl
pH 7.4 |
Other procedures may be used; for example the NANOGOLD® reagent may be used as a secondary or even tertiary labeled reagent. If additional incubation steps are used, rinse with buffer 3 (3 X 10 mins) after each incubation.
Contents
- Furuya, F. R., and Hainfeld, J. F.; J. Histochem. Cytochem., 40, 177 (1992); Furuya, F. R., Hainfeld, J. F., and Powell, R. D., Proc. 49th Ann. Mtg., Micros. Soc. Amer.; Bailey, G. W., and Hall, E. L. (Eds.); San Francisco Press, San Francisco, CA,1991, p.286.
- Lipka, J. J., Hainfeld, J. F., and Wall, J. S., J. Ultrastruct. Res., 84, 120 (1983).
- Shah, N., Zhang, S., Harada, S., Smith, R. M., and Jarrett, L.: Endocrinology, 136, 2825-2835 (1995).
- D'Souza, S. E., et al, J. Biol. Chem., 263, 3943 (1988).
- J. E. Beesley, in Colloidal Gold: Principles, Methods and Applications, M. A. Hayat, ed., Academic Press, New York, 1989; Vol. 1, pp. 421-425.
- Arai, R., et al.; Brain Res. Bull. 28, 343-345 (1992).
- Moeremans, M. et al., J. Immunol. Meth. 74, 353 (1984).
Questions? Technical Support is available. Drop us a line-- we're here to help.
|
Nanoprobes, Incorporated
95 Horseblock Road, Unit 1, Yaphank, NY 11980-9710
Tel: (877) 447-6266 (Toll-Free in US) or (631) 205-9490 | Fax: (631) 205-9493
tech@nanoprobes.com | www.nanoprobes.com |
For a complete list of references citing this product, please visit our References page.
Contents
|